ConPtm,(n+m≤7)团簇吸附CO的第一性原理研究
时间:2023-01-29 19:15:03 来源:天一资源网 本文已影响 人 
周庭艳, 张仁丽, 杨 昆, 黄海深, 吴 波
(1. 遵义师范学院 物理与电子科学学院, 遵义 563006;2. 凤凰中学, 兴仁 562300; 3. 贵州师范大学 物理与电子科学学院, 贵阳 550025)
21世纪的今天,随着人们的生活水平不断提高,环境保护成为全世界共同关注的话题. 工业和汽车排放的废气中含有大量有害的碳、氮化合物,例如CO、CO2和NO2等. 这些物质给大气带来了严重的污染,是人类健康的主要威胁之一. 含碳化合物中,CO给人类带来的危害最大. 一氧化碳是一种无味无色且有毒的气体,是含碳物质燃烧不完全的一种产物. 若将其继续燃烧则会产生能带来温室效应的气体CO2. 除此之外,CO非常容易与血红蛋白结合,形成碳氧血红蛋白,使血红蛋白丧失携氧的能力和作用,造成组织窒息,因此它对全身的组织细胞均有毒性作用,影响最为严重的是大脑皮质. 所以,降低空气中CO的含量对人类的健康有着非常重要的意义[1, 2]. CO与纳米材料之间的相互作用为控制CO在空气中的含量提供了有效途径,成为国内外学者关注的焦点[3, 4].
铂的双金属纳米颗粒具有独特的磁性和催化活性,可用于高级信息存储、电磁学、生物医学、化学催化等领域. 因此,铂的双金属纳米颗粒在过去的几十年中成为团簇研究的热点之一[5]. 其中Co-Pt纳米合金团簇,由于被认为是超高密度磁存储的最佳候选材料,更是吸引了广大学者的注意[6, 7]. 例如,Li等人[8]利用自旋极化密度泛函理论中的平面波赝势方法研究了ComPtn(m+n≤10)团簇的结构和磁性.Co原子一般位于团簇内部,而Pt原子则倾向于分布在表面附近. 富Pt团簇中Co和Pt原子上的磁矩普遍大于富Co团簇中的原子磁矩. Co-Pt纳米颗粒也具有良好的催化活性,例如Lai等人[5]的实验结果表明,Pt1Co1/C纳米催化剂的氧化还原反应活性较强,并认为这种较强的活性源于Pt原子和Co原子之间较高的合金化程度及Pt原子合适的电子结构.
CoPt团簇具有优良的磁性和催化活性,其吸附和催化CO小分子以及CO对其磁性的影响具有重要的研究价值,目前这方面的研究还比较少.本文采用密度泛函理论(DFT)研究双金属ConPtm(n+m≤7)团簇与CO小分子之间的相互作用,揭示Co、Pt原子比例对CO吸附强度的影响规律以及CO分子对CoPt团簇磁性的调控作用.
利用基于密度泛函理论的DMol3模块计算[9],结构优化和电子性质的计算采用广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof (PBE)交换关联泛函[10]. 在计算过程中,电子结构计算的判据为10-6Hartree. 以力、位移和能量作为几何结构优化的判据,力和位移收敛精度分别为0.005 Hartree/Å和0.002 Å,能量收敛精度为10-5Hartree.
由Hirshfeld布居分析获得体系的电荷,下式给出了CO分子的吸附能(Ead)[11, 12]:
Ead=E(ConPtm)+nE(CO)-E(ConPtm·CO)
式中E(ConPtm),E(CO),E(ConPtm·CO)分别为ConPtm,CO分子和ConPtm·CO的总能量,Ead大于零是放热反应,Ead小于零是吸热反应.
为了检验所用方法的正确性,我们首先计算了二聚体Co2、Pt2的键长分别为:2.05 Å、2.39 Å,与其他理论计算值(2.31 Å[13]、2.33 Å[14])基本吻合.这说明所用方法对该体系是合适的.
3.1 ConPtm·CO (n+m≤7)的几何结构
首先,我利用以上计算方法,对ConPtm(n+m≤7)团簇的所有可能异构体进行了优化. 得到了ConPtm(n+m≤7)团簇的基态结构,与其他理论计算[8]相比,结果基本一致. 然后,分两步设置CO分子吸附的初始结构. 第一,将CO的C端分别在ConPtm(n+m≤7)团簇的顶位、桥位和空穴吸附CO. 第二,将CO的O端分别在ConPtm(n+m≤7)团簇的顶位、桥位和空穴吸附CO. 对所有初始结构在GGA方案下进行几何优化和频率计算. 对于含有虚频的结构,通过结构的适当调整再进行优化,确保得到稳定结构. 对特定的尺寸,在所有稳定结构中,能量最低的为该尺寸的基态结构,其他为来稳态结构. 图1给出了我们计算的ConPtm和ConPtm·CO (n+m≤7)团簇的基态结构. 基态结构图按原子总数逐渐递增,且Co原子为降序排列,我们发现随着Pt原子的掺杂Con团簇的平面结构逐渐变成三维的.

图1 ConPtm和ConPtm·CO (n+m≤7)团簇的基态结构.Fig. 1 The ground-state structures of ConPtm and ConPtm·CO (n+m≤7) clusters.
CO分子的吸附会影响ConPtm团簇中原子间的成键. 通过分析发现,ConPtm·CO (n+m≤7)团簇的成键特点呈现出规律性变化,当n+m≤5时,CO更易与在Co原子的顶部成键. 当5≤n+m≤7时,CO则更易与Pt原子成键. 并且,n+m的值一定时,m的值越大CO就逐渐的倾向于与Pt原子成键. 如图2,给出了我们计算的ConPtm(5≤n+m≤7)和ConPtm·CO (5≤n+m≤7)团簇的基态结构.
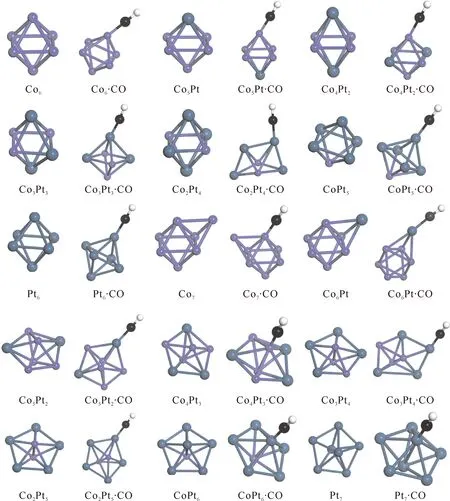
图2 ConPtm和ConPtm·CO (5≤n+m≤7)团簇的基态结构.Fig. 2 The ground-state structures of ConPtm and ConPtm·CO (5≤n+m≤7) clusters.
3.2 吸附能
通过比较能量我们发现,CO在双金属ConPtm(n+m≤7)团簇上更倾向于吸附在Pt、Co的顶部位置上. 而不是以桥位的形式吸附在两原子之间,即顶位吸附比桥位吸附和空穴吸附更具有能量优势. 桥位吸附或穴位吸附不能形成稳定的结构. 为了研究CO在双金属ConPtm(n+m≤7)团簇上的吸附强度,图3给出了CO在双金属Co-Pt上的吸附能. 我们可以看出,团簇的吸附能随着n+m的值变化而变化,但也显现一定的规律性,例如,n+m相同时,随着m的增加,吸附能增加. 即Pt原子的含量增加有利于提高团簇对CO分子的吸附强度. 其中Co2·CO、Co6·CO、Co5Pt·CO的吸附能比较小,分别为1.61 eV、1.65 eV、1.68 eV. 其余结构的吸附能比较大,基本在2.5 eV上下波动. 总的来说,Co2·CO的吸附能(1.61 eV)最低,Pt6·CO的吸附能(3.01 eV)最高,吸附能在1.61 eV到3.01 eV之间.
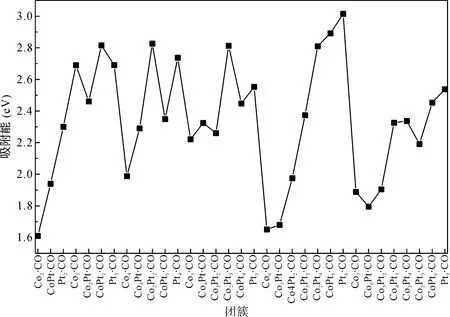
图3 CO在ConPtm (n+m≤7)基态团簇上的吸附能.Fig. 3 The adsorption energies of ConPtm·CO (n+m≤7) clusters in the ground state.
3.3 键长变化
为了讨论CO分子的吸附对ConPtm(n+m≤7)团簇结构的影响,我们计算了吸附前后原子间的距离. 图4给出了ConPtm(n+m≤7)·CO团簇中C-O键的键长随ConPtm·CO (n+m≤7)团簇尺寸增大的变化规律. 可以看出,与自由的CO分子的C-O键长(1.146 Å)相比,吸附后C-O键长均出现了明显的伸长. Co2Pt5·CO的C-O键长(1.186 Å)伸长量最大,CoPt·CO和Pt2·CO的C-O键长(1.164 Å)伸长量最小,与1.146 Å相比,伸长量分别为3%和1%. 并且C-Co的键长在1.74 Å左右,没有明显的变化. 这种吸附行为带来的键长变化应该与ConPtm(n+m≤7) 团簇的电子性质以及结构有关.
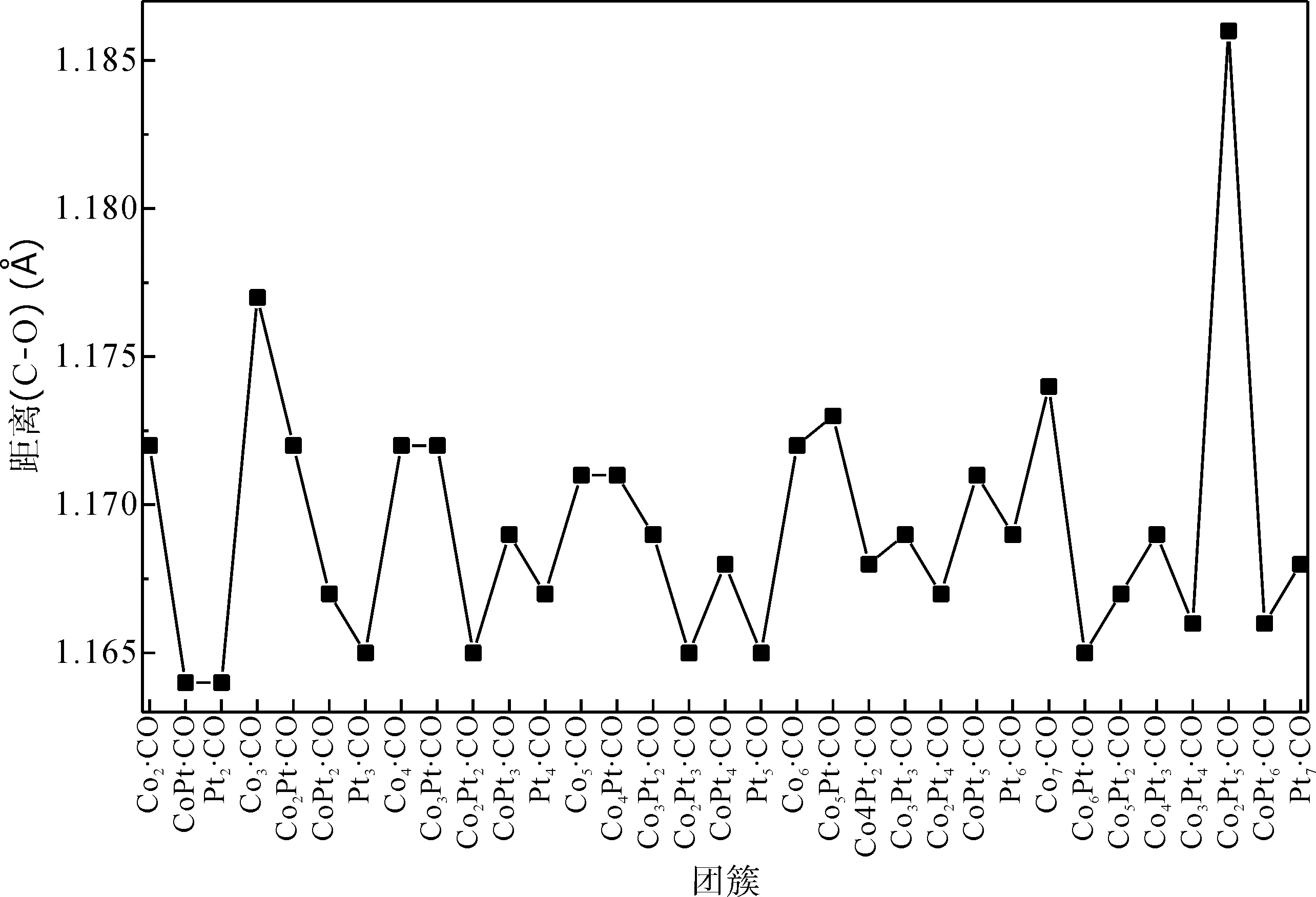
图4 ConPtm·CO (5≤n+m≤7)基态团簇中C、O原子的间距.Fig. 4 The distances between C and O of ConPtm·CO (5≤n+m≤7) clusters in the ground state.
3.4 能 隙
团簇最高占有分子轨道(HUMO)和最低未占有分子轨道(LUMO)的能级之差叫做能隙,它能够反映电子从HUMO向LUMO发生跃迁能力的大小. 普遍来讲,若能隙值越小,则相应的团簇化学活性越好. 如图5所示,我们计算给出了ConPtm·CO (n+m≤7)团簇的能隙值. 团簇ConPtm吸附CO以后能隙的变化有两种趋势. 第一,有少数团簇(如Co2Pt5、CoPt6等)由于吸附CO,其能隙值下降. 其中Pt4的能隙值减少最多,减少了0.54 eV. 第二,大部分团簇在吸附CO以后其能隙值都出现了上升,其中Co2上升的值最大,上升了0.6 eV. 这说明这部分团簇(如CoPt2等)的化学活性在吸附CO以后下降了.
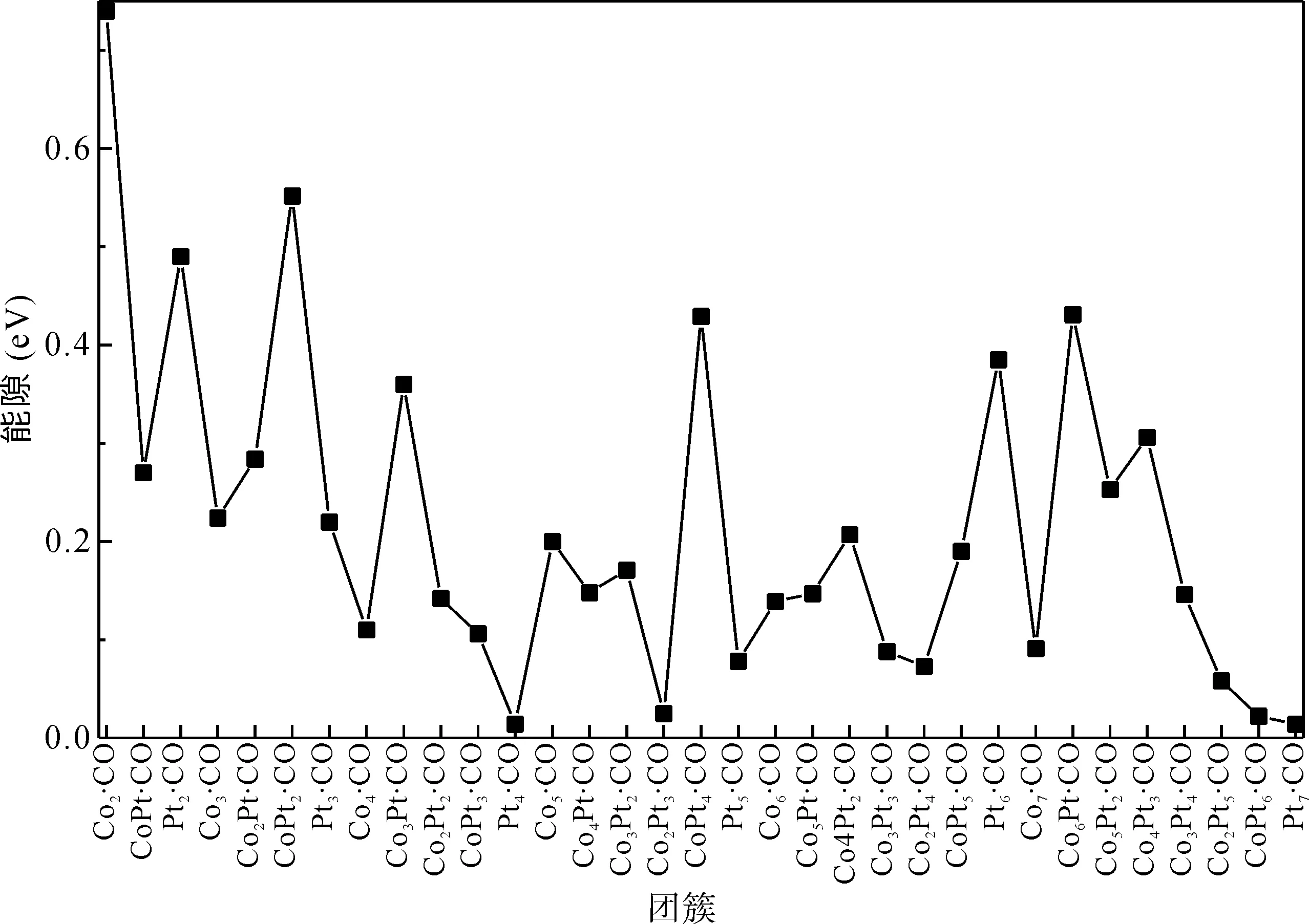
图5 ConPtm·CO (n+m≤7)基态团簇的能隙.Fig. 5 The energy gaps of ConPtm·CO (n+m≤7) clusters in the ground state.
3.5 Mulliken布局分析及磁性
轨道的电子占据数由Mulliken布局分析得到,自旋向上态与自选向下态的电子占据数之差获得磁矩. 图6给出了吸附CO前后基态团簇的总磁矩. 可以看出,Co原子的含量对磁矩影响较大,n+m相同时,n越大,团簇的磁矩越大. Pt原子的影响则相对较小. 吸附CO以后,除了Co7·CO、Co6Pt·CO、Co3Pt2·CO以及Co2Pt2·CO团簇的总磁矩下降以外,其余团簇的总磁矩均有不同程度的升高. 在CoPt·CO团簇中,所有电子两两配对,总磁矩为零,即发生了“磁矩猝灭”现象.
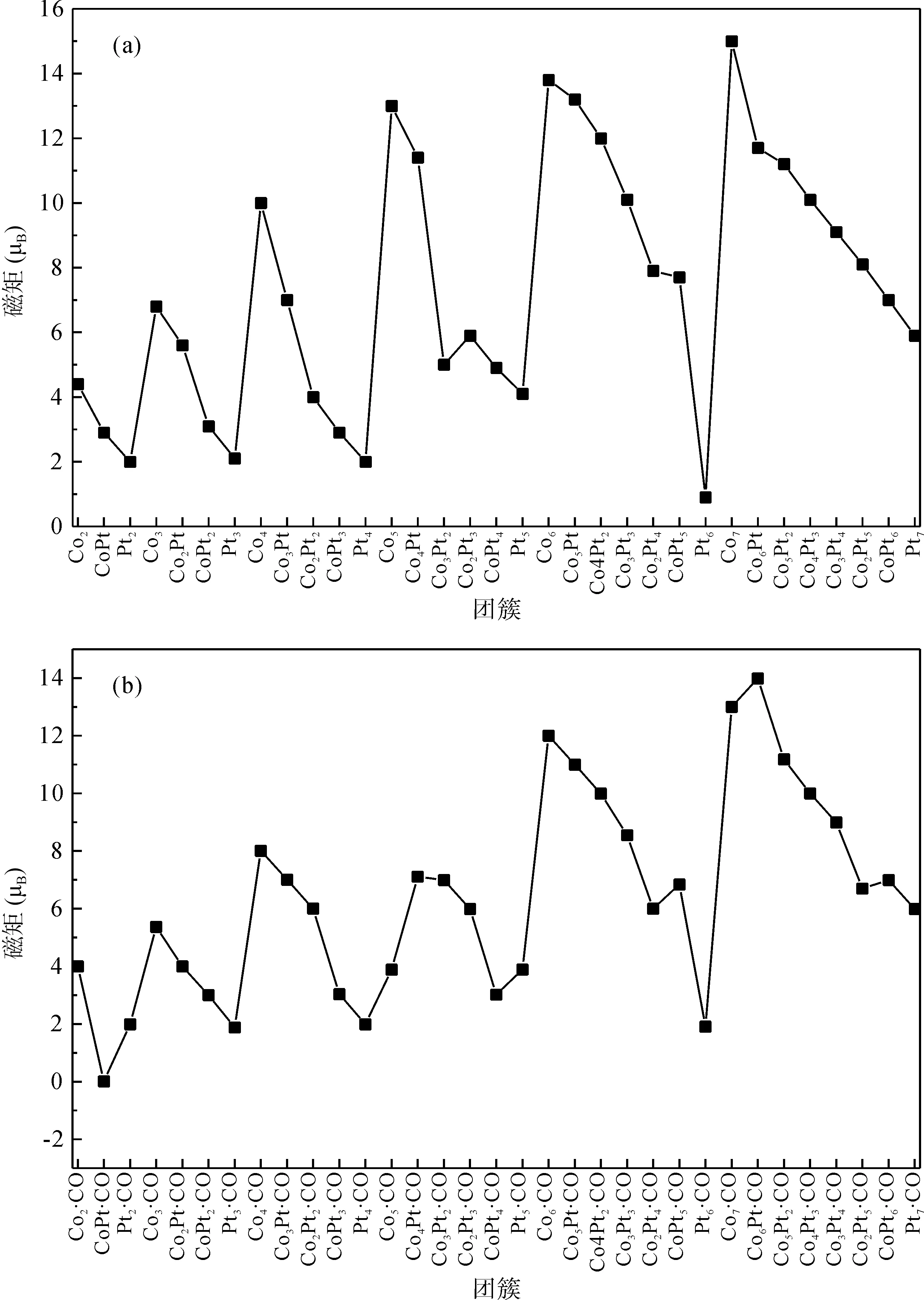
图6 (a) ConPtm和(b) ConPtm·CO (n+m≤7)基态团簇的磁矩.Fig. 6 The magnetic moments of (a) ConPtm and (b) ConPtm·CO (n+m≤7) clusters in the ground state.
本文采用密度泛函理论的第一性原理计算,研究了ConPtm(n+m≤7)团簇与CO分子之间的相互作用. 分析了CO分子的平均吸附能、键长变化以及磁性变化. 结果表明:吸附能的值大于等于1.61 eV以上. 吸附后,CO分子的C-O键长间距有所伸长,CO分子被活化. 成键特性表明,多数尺寸下,C原子倾向于与Co原子成键. CO的吸附使ConPtm(n+m≤7)团簇的总磁矩增大,并且使ConPtm(n+m≤7)团簇的能隙增大,化学活性降低.
猜你喜欢 磁性原子分子 《分子催化》征稿启事分子催化(2022年1期)2022-11-02原子究竟有多小?少儿科学周刊·儿童版(2021年22期)2021-12-11原子可以结合吗?少儿科学周刊·儿童版(2021年22期)2021-12-11带你认识原子少儿科学周刊·儿童版(2021年22期)2021-12-11围棋棋子分离器发明与创新·小学生(2019年11期)2019-08-11“精日”分子到底是什么?新民周刊(2018年8期)2018-03-02米和米中的危险分子饮食科学(2017年12期)2018-01-02自制磁性螺丝刀军事文摘·科学少年(2017年4期)2017-06-20方便磁性工具背心中学科技(2015年6期)2015-08-08臭氧分子如是说少儿科学周刊·少年版(2015年1期)2015-07-07 相关关键词: 吸附 辽宁吸附材料项目实施方案 吸附剂